


CRISPR 유전체 편집 솔루션CRISPR Genome Editing Solutions
VectorBuilders는 고객의 in vivo 및 in vitro 유전체 편집 실험을 위한 이상적인 툴들로 이용될 수 있는 포괄적인 CRISPR 제품 및 서비스를 제공합니다. 당사의 CRISPR 제품들은 높은 타겟팅 효율을 가진 transfection 또는 transduction을 위하여 바로 사용될 수 있는 제품부터 고객 맞춤형 CRISPR 벡터까지 포함하여, plasmid, 바이러스, RNA 와 같이 다양한 형식의 전달체로 제공됩니다. 고객의 CRISPR 구성요소들을 transfection이 어려운 세포들에 효율적으로 전달할 수 있도록 모든 주요 바이러스 유형 (렌티바이러스, AAV, 아데노바이러스) 으로 패키징을 할 수 있습니다. 또한, 당사는 유전자 knockout, 유전자 발현 활성화, 유전자 발현 억제, 그리고 다른 종류의 CRISPR screening을 위한 고품질의 CRISPR library 제작에 전문화되어 있습니다. 당사의 고유한 디자인으로 제작되고 잘 검증된 whole genome dual-gRNA knockout library 들은 인간과 마우스에서의 유전자들의 functional screening을 위한 강력한 도구를 제공합니다.
Click to view user testimonials about our CRISPR genome editing services
중점 사항
- 쉽고 빠른 CRISPR 디자인을 위하여 전체 유전체의 gRNA database와 함께 제공되는 매우 직관적인 온라인 벡터 디자인 플랫폼
- 풍부한 벡터 backbone 및 벡터 구성 요소 collection
- 다양한 형식의 전달체 (CRISPR/Cas9 plasmid, CRISPR/Cas9 바이러스, Cas9 mRNA + gRNA, Cas9-gRNA RNP complex 등) 로 제공되는 CRISPR 구성 요소들
- 다양한 CRISPR library 제작 옵션
- 확실한 품질, 짧은 소요 시간 및 경쟁력 있는 가격
- 실험 디자인, 데이터 분석, 문제 해결을 위한 강력한 기술 지원
제품 및 서비스 세부 사항
- 맞춤형 CRISPR 벡터
- 많이 사용되는 CRISPR 벡터
- CRISPR 바이러스
- Cas9 mRNA 및 gRNA를 위한 RNA 제조
- 정밀한 유전체 편집을 위한 donor DNA
- 혼합 CRISPR library
- CRISPR 솔루션
기술적인 정보
- CRISPR 기반의 유전체 편집
- CRISPR-매개 유전자 조절
- CRISPR 전달 방법들
- gRNA databases
제품 및 서비스 세부 사항
맞춤형 CRISPR 벡터
CRISPR 기반의 유전체 편집을 위해서는 Cas9과 타겟 부위에 결합하는 gRNA의 동시 발현이 요구됩니다. 당사의 매우 직관적인 벡터 디자인 플랫폼을 이용하여 Cas9과 고객의 gRNA를 여러 가지 다양한 방법으로 발현할 수 있도록, 30가지 이상의 벡터 backbone (non-viral, viral 또는 transposon) 과 무한한 벡터 구성 요소들 (promoters, Cas9 variants, fluorescent 및 drug-selection markers) 의 조합을 선택할 수 있습니다. 또한, 일반 plasmid, 렌티바이러스, AAV, 아데노바이러스, 또는 PiggyBac backbone을 사용하여 Cas9과 gRNA를 단일 벡터 또는 이중 벡터 시스템으로 제공합니다.
당사의 포괄적인 CRISPR 벡터 collection은 HDR에 의하여 타겟 부위에 정교한 point mutation이나 큰 서열을 knockin하는데 가이드 DNA template로 사용될 수 있는 유전자 타겟팅 donor 벡터도 포함되어 있습니다. 또한, 타겟 유전자 내부의 부위를 이용하여 전사를 활성화하거나 억제하는 CRISPRa 및 CRISPRi 벡터도 제공합니다. CRISPR에 의한 유전자 발현 활성화를 위하여 dCas9-SAM 또는 dCas9-SunTag 기반의 벡터를, CRISPR에 의한 유전자 발현 억제를 위하여 dCas9-KRAB 또는 dCas9-KRAB-MeCP2 기반의 벡터를 디자인하고 제작할 수 있습니다.
CRISPR 벡터를 shopping cart에 추가할 때 downstream 서비스로서 plasmid DNA 정제, RNA 정제 및 바이러스 패키징 등의 서비스를 구입할 수 있습니다.
디자인을 위한 팁
Note: AAV의 수용 능력은 4.7 kb입니다. 일반적으로 사용되는 Streptococcus pyogenes에서 유래된 SpCas9는 4.2 kb이며, promoter, polyA signal 서열 및 gRNA 발현 카세트를 결합하면 수용 한계를 초과하게 됩니다. 따라서 당사의 표준 AAV CRISPR 시스템은 길이가 더 짧은 Staphylococcus aureus에서 유래된 SaCas9 (3.2 kb)을 사용합니다. SpCas9에 의해 인지되는 PAM은 NGG이지만, SaCas9에 의해 인지되는 PAM은 NNGRR 또는 NNGRRT (선호됨) 라는 점에 유의하시기 바랍니다. 요청시에 AAV SpCas9 벡터도 제작해드릴 수 있습니다.
맞춤형 벡터를 선택하시기 바랍니다 View more
많이 사용되는 CRISPR 벡터
맞춤형 CRISPR/Cas9 벡터 이외에 VectorBuilder는 CRISPRa and CRISPRi를 위한 premade Cas9 벡터, control gRNA 벡터 및 helper 벡터의 panel을 제공합니다. 바로 사용할 수 있는 벡터들은 주문 즉시 배송 가능합니다. Premade 벡터를 shopping cart에 추가할 때 downstream 서비스로서 plasmid DNA 정제, RNA 정제 및 바이러스 패키징 등의 서비스를 구입할 수 있습니다.
당사의 premade CRISPR 벡터 collection을 보시려면 여기를 눌러주시기 바랍니다. View more
| Vector System | Vector Name | Vector ID |
|---|---|---|
| hCas9 expression vector (regular plasmid) | pRP[Exp]- mCherry/Hygro-CBh>hCas9 |
VB010000-9378bvk |
| hCas9 expression vector (lentivirus) | pLV[Exp]- CBh>hCas9/Hygro |
VB010000-9380sne |
| SaCas9 expression vector (AAV) | pAAV[Exp]-CMV>SaCas9 | VB010000-9382per |
| hCas9 expression vector (adenovirus) | pAV[Exp]-CBh>hCas9 | VB010000-9381pwj |
| hCas9 expression vector (piggyBac) | pPB[Exp]-mCherry/Hygro-CBh>hCas9 | VB010000-9379gqq |
| Scramble gRNA control vector (regular plasmid) | pRP[gRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9358ttk |
| Scramble gRNA control vector (lentivirus) | pLV[gRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9359hhe |
| Scramble SagRNA control vector (AAV) | pAAV[SagRNA]-EGFP-U6>Scramble_SagRNA1 | VB010000-9361zjr |
| Scramble gRNA control vector (adenovirus) | pAV[gRNA]-EGFP-U6>Scramble_gRNA1 | VB010000-9360nph |
| Scramble gRNA control vector (piggyBac) | pPB[gRNA]-EGFP/Puro-U6>Scramble_gRNA1 | VB010000-9362zuk |
| hCas9 and scramble gRNA coexpression vector (regular plasmid) | pRP[CRISPR]-EGFP/Puro-hCas9-U6>Scramble_gRNA1 | VB010000-9354ztt |
| hCas9 and scramble gRNA coexpression vector (lentivirus) | pLV[CRISPR]-hCas9/Puro-U6>Scramble_gRNA1 | VB010000-9355sqw |
| SaCas9 and scramble SagRNA coexpression vector (AAV) | pAAV[CRISPR]-SaCas9-U6>Scramble_SagRNA | VB010000-9357zmm |
| hCas9 and scramble gRNA coexpression vector (adenovirus) | pAV[CRISPR]-hCas9/EGFP-U6>Scramble_gRNA1 | VB010000-9356pna |
| dCas9-SAM activator MS2/P65/HSF1 expression vector (lentivirus) | pLV[Exp]-EF1A>MS2/P65/HSF1/Hygro | VB010000-9383ffr |
| dCas9-SAM activator dCas9/VP64 expression vector (lentivirus) | pLV[Exp]-EF1A>dCas9/VP64/Bsd | VB010000-9384wwc |
| dCas9-SAM scramble msgRNA control vector (lentivirus) | pLV[msgRNA]-EGFP/Puro-U6>Scramble_gRNA | VB010000-9363gsm |
| dCas9-KRAB-MeCP2 expression vector (lentivirus) | pLV[Exp]-CBh>dCas9/ KRAB/MeCP2/Hygro |
VB010000-9386mwf |
CRISPR 바이러스
렌티바이러스, AAV 및 아데노바이러스 는 포유류 세포에 CRISPR 구성 요소들을 전달하기 위하여 폭넓게 사용되고 있습니다. VectorBuilder는 transfection이 어려운 세포들에 매우 높은 효율의 CRISPR 타겟팅이 가능하도록 렌티바이러스, AAV, 아데노바이러스에 대한 프리미엄급 바이러스 패키징 서비스를 제공합니다. 당사 자체의 기술과 시약으로 titer, 순도, viability, 일관성이 크게 향상된 바이러스 패키징 방법을 개발해왔습니다. 당사의 패키징 방법은 벡터 제작 서비스에서 사용되는 바이러스 벡터 시스템들을 위해서도 최적화되었습니다. 그 결과로, 클로닝과 바이러스 패키징에 대한 고객의 수요를 만족시켜서 당사에 이들 서비스를 다시 의뢰하는 고객이 점차로 증가하고 있습니다.
렌티바이러스 패키징의 가격, 소요 시간 및 규모View more
| Scale | Application | Typical Titer | Minimum Titer | Volume | Price (USD) | Turnaround |
|---|---|---|---|---|---|---|
| Pilot | Cell culture | >4x108 TU/ml | >108 TU/ml | 250 ul (10x25 ul) | $449 | 6-12 days |
| Medium | >3x108 TU/ml | 1 ml (10x100 ul) | $649 | |||
| Large | >2x109 TU/ml | >109 TU/ml | 1 ml (10x100 ul) | $1,099 | ||
| Ultra-purified medium | Cell culture & in vivo | >2x109 TU/ml | >109 TU/ml | 500 ul (10x50 ul) | $1,399 | |
| Ultra-purified large | 1 ml (10x 100 ul) | $1,699 |
TU = Transduction units (also known as infectious units)
AAV 패키징의 가격, 소요 시간 및 규모View more
| Scale | Application | Typical Titer | Minimum Titer | Volume | Price (USD) | Turnaround |
|---|---|---|---|---|---|---|
| Pilot | Cell culture | >1012 GC/ml | >2x1011 GC/ml | 250 ul (10x25 ul) | $449 | 6-12 days |
| Medium | 1 ml (10x100 ul) | $649 | ||||
| Large | >5x1012 GC/ml | >2x1012 GC/ml | 1 ml (10x100 ul) | $1,099 | ||
| Ultra-purified pilot | Cell culture & in vivo | >2x1013 GC/ml | >1013 GC/ml | 100 ul (4x25 ul) | $1,399 | 7-14 days |
| Ultra-purified medium | 500 ul (10x50 ul) | $1,999 | ||||
| Ultra-purified large | 1 ml (10x100 ul) | $3,099 |
GC = Genome copies
아데노바이러스 패키징의 가격, 소요 시간 및 규모View more
| Scale | Application | Typical Titer | Minimum Titer | Volume | Price (USD) | Turnaround |
|---|---|---|---|---|---|---|
| Pilot | Cell culture | >2x1010 IFU/ml | >1010 IFU/ml | 250 ul (10x25 ul) | $649 | 28-35 days |
| Medium | 1 ml (10x100 ul) | $1,099 | ||||
| Large | >2x1011 IFU/ml | >1011 IFU/ml | 1 ml (10x100 ul) | $1,699 | ||
| Ultra-purified medium | Cell culture & in vivo | >2x1012 VP/ml | >1012 VP/ml | 500 ul (10x50 ul) | $2,099 | |
| Ultra-purified large | 1 ml (10x100 ul) | $2,499 |
IFU = Infectious units; VP = Virus particles
Cas9 mRNA 및 gRNA를 위한 RNA 제조
VectorBuilder는 고객이 원하시는 타겟 부위에 대하여 디자인된 CRISPR 구성 요소들을 RNA 형태로 포유류 세포에 용이하게 전달할 수 있는 Cas9 mRNA 및 gRNA를 바로 transfection이나 micro injection에 사용할 수 있도록 제조해드립니다. Cas9 mRNA는 야생형 hCas9 nuclease와 Cas9 nickase (Cas9(D10A))로 제공해드릴 수 있습니다. 우리는 CRISPR library design(CLD)에 활용된 알고리즘으로 특이성 점수를 계산하고 일련의 경험적 규칙을 적용하여 사용자가 선택한 유전자/사이트를 타겟팅하기 위한 최적의 gRNA를 디자인합니다.
CRISPR RNA 제조 가격 및 소요 시간 View more
| Reagent | Concentration & Volume | Price (USD) | Turnaround |
|---|---|---|---|
| hCas9 mRNA | >500 ng/ul, 25 ul, nuclease-free water, sterile | $449 | 2-4 days |
| Cas9(D10A) mRNA | |||
| Custom gRNA* | $349 |
*gRNA를 in vitro transcription 벡터로 클로닝하기 위해서는 $149의 추가 비용과 5-10 일의 소요 시간이 요구됩니다.
정밀한 유전체 편집을 위한 donor DNA
VectorBuilder는 CRISPR 절단 부위의 정교한 DNA 서열 변화를 도입하기 위하여 HDR에 의한 RNA repair를 가이드하는 donor DNA template를 ssODN (single-stranded oligodeoxynucleotides) 또는 plasmid를 선형화하여 만든 dsDNA 로 제공해드립니다. 도입될 수 있는 서열 변화는 point mutation이나 작거나 큰 DNA 서열의 knockin을 포함합니다. ssODN은 작은 tag이나 제한효소 부위와 같은 60 bp 미만의 짧은 DNA 서열을 삽입하는데 사용되며, dsDNA donor는 형광 tag이나 다른 reporter와 같이 4-5 kb까지의 더 큰 서열의 targeted knockin을 위하여 사용됩니다.
Donor DNA 제조 가격 및 소요 시간View more
| Reagent | Price (USD) | Turnaround |
|---|---|---|
| ssODN (normally 120-200 nt) | From $349 | 2-3 weeks |
혼합 CRISPR library
VectorBuilder는 유전자의 functional screening을 위한 knockout library, 유전자의 gain-of-function screening 또는 noncoding 서열의 regularory function screening을 위한 CRISPRa library, 유전자의 loss-of-function screening 또는 noncoding 서열의 regularory function screening을 위한 CRISPRi library 등과 같이 CRISPR 응용 분야에 일반적으로 사용되는 다양한 혼합 library를 맞춤형으로 디자인하고 제작하는 전문성을 가지고 있습니다. 또한, single cell 기반의 screening이나 CRISPR 기술을 사용하는 다른 혼합 library를 위한 CRISPR barcode library도 제작 가능합니다.
고객의 library는 E. coli stock, plasmid DNA pool, 또는 패키징된 바이러스 형태로 보내드립니다. 당사의 맞춤형 library는 NGS에 의하여 완전히 검증이 되기 때문에, 어떤 library를 받게 되는지 바로 아실 수 있습니다.
당사의 library 제작 서비스에 대한 상세한 정보를 원하시면 여기를 눌러주시기 바랍니다.
맞춤형 혼합 CRISPR library 이외에 인간과 마우스에 대한 whole-genome knockout screening에 사용될 수 있는 premade 이중 gRNA 렌티바이러스 library 도 제공합니다. 이중 gRNA library는 하나의 유전자를 타겟팅하는 두 개의 gRNA에 의하여 큰 deletion을 도입할 수 있어서 단일 gRNA에 비하여 loss-of-function을 초래하는 변이를 생성하는데 있어서 더 효율적이기 때문에 knockout screening을 위하여 훨씬 더 효율적입니다. 20,048개의 인간 유전자와 20,493개의 마우스 유전자를 타겟팅할 수 있는 이중 gRNA library들이 즉시 사용할 수 있는 높은 titer의 혼합 렌티바이러스 형태로 만들어집니다. 가능한 경우, 각각의 유전자는 중복된 4-6개의 서로 다른 gRNA 쌍에 의하여 타겟팅됩니다.
Premade dual-gRNA 제조 가격 및 소요 시간View more
| Product name | No. of genes | No. of gRNA pairs | Scale | Catalog No. | Price (USD) |
|---|---|---|---|---|---|
| Human Whole-Genome Dual-gRNA Lentivirus Library | 20,048 | 91,926 | Medium (>1.0x108 TU/ml, 1 ml) |
LVM(Lib190505-1046fgb) |
$2,999
|
| Plus (>1.0x108 TU/ml, 5 ml) |
LVM(Lib190505-1046fgb) |
$3,499
|
|||
| Mouse Whole-Genome Dual-gRNA Lentivirus Library | 20,493 | 90,344 | Medium (>1.0x108 TU/ml, 1 ml) |
LVM(Lib190505-1050kpm) |
$2,999
|
| Plus (>1.0x108 TU/ml, 5 ml) |
LVM(Lib190505-1050kpm) |
$3,499
|
당사의 premade 이중 gRNA CRISPR library에 대한 상세한 정보를 원하시면 여기를 눌러주시기 바랍니다.
CRISPR 솔루션
CRISPR 시스템의 다양성은 포유류 세포에서 폭넓은 유전체 편집 목적을 위한 적합성을 제공합니다. 당사의 분자생물학 응용 분야에 광범위한 경험을 가진 기술팀이 실험 디자인부터 바로 사용할 수 있는 시약 제조까지 고객의 CRISPR 유전체 편집 프로젝트를 위한 완전한 솔루션을 제공해드립니다. 아래의 리스트와 같은 일반적인 CRISPR 응용 분야를 위해서, 또는 고객의 새로운 CRISPR 응용 분야를 위해서 VectorBuilder가 함께 해드립니다. 무료 서비스 제안서를 받기를 원하시면 오늘 바로 디자인 의뢰하기 를 통하여 문의해주시기 바랍니다!
-
점 변이 (point mutation) 도입
-
Fragment knockin
- Small tag 삽입 (< 60 bp)
- Large fragment 삽입 (4-5 kb 까지)
-
CRISPR 유전자 발현 활성화
-
CRISPR 유전자 발현 억제
-
CRISPR library (KO, CRISPRa, CRISPRi, barcoding, 등.)
-
안정한 세포주 제작
- Cas9을 발현하는 세포주
- iPSC 유전체 편집
- 암세포 유전체 편집
기술적인 정보
CRISPR 기반의 유전체 편집
기존의 CRISPR 시스템은 두가지 구성요소를 포함합니다: Cas9 단백질 및 guide RNA (gRNA). 가장 일반적으로 사용되는 Cas9은 Streptococcus pyogenes (일명 SpCas9 또는 hCas9) 에서 유래되어 만들어졌습니다. Cas9은 RNA-guided DNA nuclease로 타겟 부위에 double-stranded breaks (DSBs) 를 생성합니다 (Figure 1). 또 다른 널리 이용되는 변형 Cas9으로 Cas9 “nickase” (예를 들면, Cas9(D10A))가 있으며, 이는 DNA에 single-stranded cut을 생성합니다. Cas9 nickase를 타겟 부위 주위의 두 상반된 strand를 타겟팅하는 gRNA들과 함께 사용하면, nickase가 각각의 strand에 하나씩의 절단 부위를 생성하여 결과적으로 타겟 부위에 DSB가 생성됩니다. CRISPR 시스템에 의하여 DSB가 형성되면, 세포의 non-homologous end joining (NHEJ) pathway가 활성화되어 DNA 절단 부위를 수리하게 되는데, 그 결과로 일반적으로 small random deletion이 생기며 드물게 insertion이나 base substitution이 일어나기도 합니다. 이러한 변이 (예를 들어, frameshift가 일어나게 하는 deletion) 가 단백질을 코딩하는 영역에서 생기면, 유전자의 기능적인 knockout이 일어나게 됩니다.

Figure 1. CRISPR-induced DNA repair의 메커니즘.
덜 효율적이지만 또 다른 방법으로는 외래의 donor DNA template를 CRISPR 구성 요소들과 함께 도입하는 경우에 DSB가 homology-directed repair (HDR) pathway에 의하여 수리되기도 합니다. 이 경우에는 유전체 내의 타겟 서열이 donor 서열과 교체되어 point mutation 또는 donor DNA 서열의 knockin과 같은 정교한 서열 변화를 가능하게 합니다. Donor DNA template로는 single-stranded oligo nucleotide (ssODN) 또는 dsDNA fragment (대개는 선형화된 plasmid DNA) 가 사용될 수 있습니다. ssODN은 point mutation이나 small tag 삽입에 적합하고, dsDNA fragment는 large fragment knockin을 위해서 널리 이용됩니다.

Figure 2. gRNA-Cas9 ribonucleoprotein (RNP) 방법을 사용하여 homozygous CD274 knockout (KO) 돌연변이 생성. (A) 편집 RNP는 타겟 세포에 electroporation되고 단일 클론이 분리하고 스크리닝합니다. 후보의 유전자형은 PCR 및 Sanger 시퀀싱을 사용하여 검증합니다. (B) murine colon adenocarcinoma cell line을 편집하는 이 사례 연구에서, 13-kbp를 KO하기 위해 타겟 유전자의 2개 부위에 결합하는 RNP로 세포를 electroporation하였습니다. 4개의 프라이머인 P1에서 P4는 KO 및 WT 클론을 구별하기 위해 세번의 PCR에 사용되었습니다. (C) PCR 결과를 기반으로 클론 1은 homozygous KO 돌연변이인 것으로 확인되었으며, 이는 (D) 시퀀싱 결과로도 확인됩니다.
CRISPR-매개 유전자 조절
촉매 반응으로 불활성인 Cas9 형태(일명 dCas9)는 내인성 게놈 유전자좌의 CRISPR 매개 transcription 조절을 달성하기 위해 다른 transcription 복합체와 결합될 수 있습니다. 유전자 transcription을 활성화하기 위해 SAM(Synergistic Activation Mediator) 시스템을 사용할 수 있습니다. 이 시스템은 dCas9/VP64 fusion protein, MS2/P65/HSF1 helper 및 modified gRNA 세가지 구성 요소를 사용합니다. dCas9 단백질은 합성 transcriptional activator인 VP64와 변형 조작되며, gRNA는 NF-kB(p65) 및 HSF1의 transactivation 도메인으로 구성된 MS2 복합체를 모집하는 MS2 aptamers를 포함하도록 변형됩니다. VP64는 herpes simplex virus protein 16의 4개의 tandem repeats으로 구성되며 Cas9 단백질의 C-말단에 융합되어 유전자 발현을 활성화하는 전사 기구를 모집하게 됩니다. p65 및 HSF1의 transactivation 도메인은 promoter 또는 enhancer에 transcription을 관여하는 cofactors를 추가로 모집하는 역할을 하는 강력한 transcriptional activator입니다. 또한 dCas9/VP64 및 MS2/P65/HSF1 복합체는 Figure 3에서와 같이 타겟 유전자 발현을 활성화하기 위해 상승적으로 작동합니다. SAM 시스템 외에도 VectorBuilder는 HSF1의 transactivation 도메인을 Epstein-Barr virus R transactivator로 대체한 dCas9-VP64-p65-Rta(VPR) transactivation 도메인 시스템을 제공합니다.
반면, 타겟 유전자 transcription을 억제하기 위해 dCas9는 KRAB(Kruppel-associated box domain) 및 MeCP2와 같은 transcriptional repressor와 변형 조작될 수 있습니다. dCas9/KRAB helper 벡터의 두가지 버전이 현재 사용 가능합니다. 오리지날 dCas9/KRAB helper 벡터 버전과 DNA 타겟 부위의 더 강력한 전사 억제를 달성하기 위해 bipartite repressor 도메인인 KRAB/MeCP2에 융합된 dCas9의 발현을 유도하는 개선된 dCas9/KRAB/MeCP2 helper 벡터 버전입니다. Figure 4과 같이 gRNA가 dCas9/KRAB/MeCP2를 내인성 promoter로 모집하면 transcription이 억제됩니다. KRAB 도메인은 human transcription factor에서 일반적으로 발견되며 현재 인간 게놈에서 확인된 가장 강력한 transcriptional repressor 중 하나입니다. MeCP2는 histone deacetylase의 모집을 통해 KRAB 매개 억제를 더욱 강화하여 chromatin condensation 및 epigenetic-mediated repression을 초래합니다. CRISPR 매개 유전자 조절은 chromatin에 대한 regulatory element 테스트, 타겟 유전자 조절 또는 whole genome screening을 포함한 다양한 응용 분야에 사용할 수 있습니다.
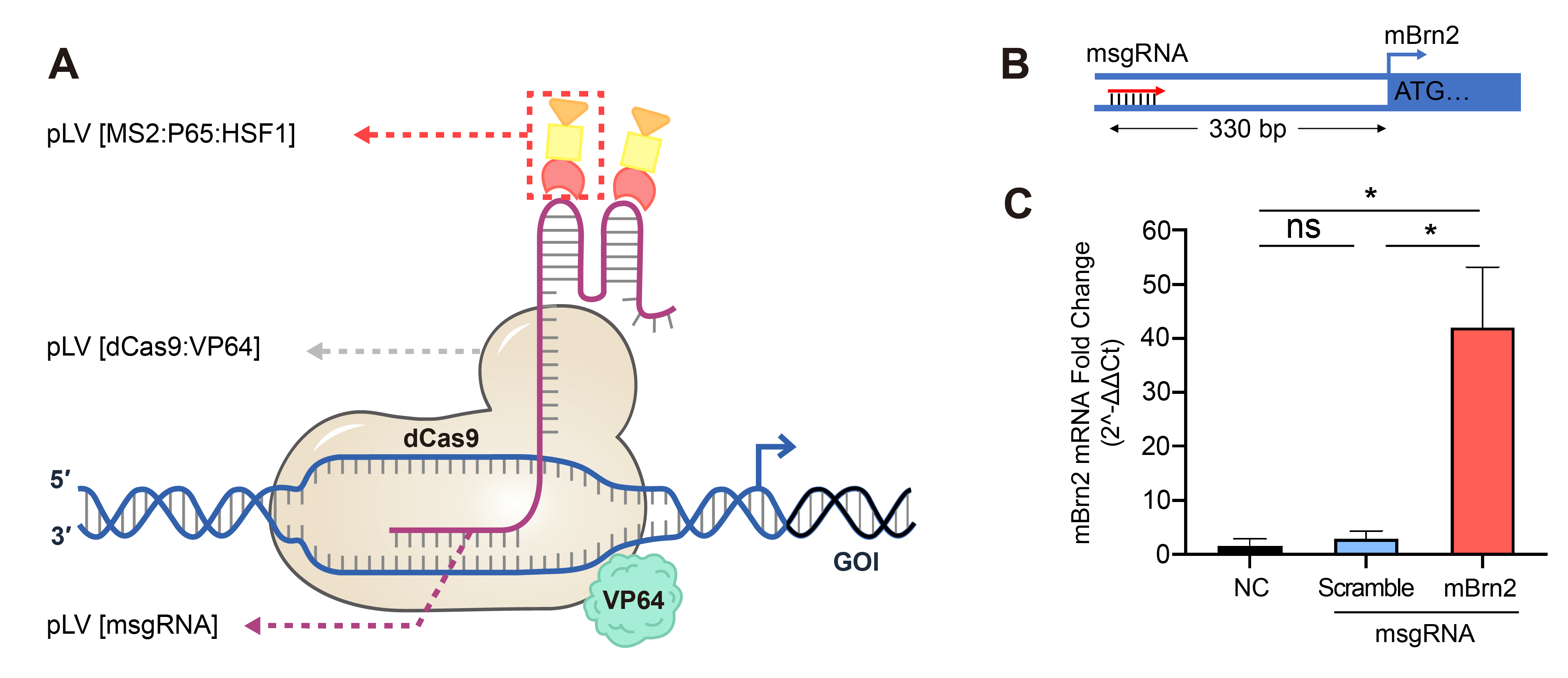
Figure 3. Lentivirus-based CRISPRa에 의해 달성된 유전자 발현의 상향 조절. SAM 복합체인 dCas9/VP64 및 MS2/P65/HSF1을 안정적으로 발현하는 NIH3T3 세포에 msgRNA 발현 렌티바이러스를 transduction한 후 항생제로 선별하였습니다. (A) 전사 활성화를 조절하는 SAM 시스템. (B) 마우스 Brn2 유전자의 프로모터 영역을 타겟으로 하는 msgRNA 디자인. (C) qRT-PCR에 의해 측정된, scramble 또는 타겟 msgRNA 또는 무처리 대조군(NC)으로 transduction된 NIH3T3 세포에서 Brn2의 상대적 유전자 발현. Mean±SD, *P<0.05, ANOVA with Tukey’s post hoc test.
.png")
Figure 4. Lentivirus-based CRISPRi에 의해 달성된 유전자 발현의 하향 조절. dCas9/KRAB/MeCP2 transcriptional repressor 복합체를 안정적으로 발현하는 Jurkat 세포를 gRNA 발현 렌티바이러스로 transduction한 후 항생제로 선별하였습니다. (A) 유전자 전사 억제를 조절하는 dCas9/KRAB/MeCP2. (B) Human CXCR4 유전자의 프로모터 영역을 타겟으로 하는 gRNA 디자인. (C) Western blot에 의해 측정된 scramble 또는 타겟 gRNA 또는 무처리 대조군(NC)으로 transduction된 Jurkat 세포의 CXCR4 단백질 레벨. (D) qRT-PCR에 의해 측정된 scramble 또는 타겟 gRNA 또는 무처리 대조군(NC)으로 transduction된 Jurkat 세포에서의 상대적 CXCR4 유전자 발현. Mean±SD, ***P<0.001, ****P<0.0001, ANOVA with Tukey’s post hoc test. (E) Scramble 또는 타겟 gRNA로 transduction된 Jurkat 세포에서 표면 발현 CXCR4를 flowcytometry에 의해 정량화하였습니다. CXCR4는 단클론 1차 항체(Ab) 및 fluorophore-conjugated 2차 Ab로 표지 되었습니다. 표지되지 않은 것과 2차 Ab. Jurkat 세포만 음성 대조군으로 사용하였습니다. (F) CXCR4 타겟 gRNA로 transduction된 세포 표면의 CXCR4 양은 scramble gRNA로 transduction된 세포에 비해 평균적으로 약 50% 감소하였습니다. Mean±SD.
CRISPR 전달 방법들
CRISPR 구성 요소들은 다음과 같은 다양한 방법으로 포유류 세포 내로 도입될 수 있습니다 (Figure 5):
- gRNA 및 Cas9 plasmid
- gRNA 및 Cas9 바이러스 (렌티바이러스, AAV, 아데노바이러스 등.)
- gRNA와 Cas9 mRNA 혼합물
- gRNA와 Cas9 단백질의 preformed RNP
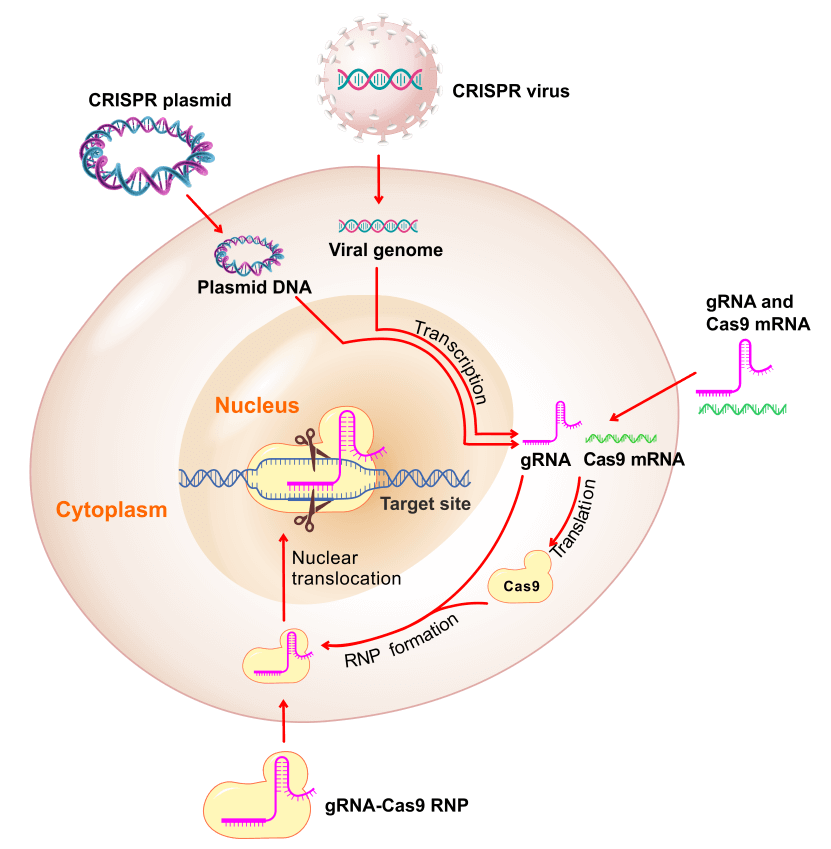
Figure 5. CRISPR 구성 요소들을 세포로 전달하는 방법들.
아래의 표에는 각각의 전달 방법에 대한 중요한 장단점이 요약되어 있으며, 귀하의 실험을 위한 가장 적합한 전달 방법을 결정할 때 reference로 사용될 수 있습니다:
| 전달 방법 | 장점 | 단점 |
|---|---|---|
| gRNA 및 Cas9 plasmid |
|
|
| gRNA 및 Cas9 바이러스 |
|
|
| gRNA와 Cas9 mRNA 혼합물 |
|
|
| gRNA와 Cas9 단백질의 preformed RNP complex |
|
|
gRNA databases
VectorBuilder의 온라인 CRISPR 벡터 디자인 툴은 인간, 마우스, 쥐에서 높은 타겟팅 효율을 갖는 CRISPR 벡터를 디자인 할 수 있도록 하는 최적화된 whole-genome gRNA database를 특징으로 갖추고 있습니다. 우리는 CRISPR library design(CLD)에 활용된 알고리즘을 따라 gRNA에 대한 특이성 점수를 계산합니다. 간략하게, 하나의 생물종에 대한 주어진 gRNA가 N(20)NGG 서열을 타켓팅한다고 할 때, 그 생물종의 유전체 서열 내에서 타켓 서열과 3개 이하의 mismatch를 갖는 모든 잠재적인 off-target 서열들을 검색하고, 각각의 잠재적인 off-target 부위에 대하여 off-target score를 계산합니다. 모든 off-target 부위의 score를 종합하여 최종적인specificity score를 계산하며, 이 score는 0과 100 사이의 값으로 score가 더 클수록 더 높은 targeting specificity를 의미합니다. Specificity score는 대략적인 가이드에 불과하다는 점에 유의하시기 바랍니다. 실제 타켓팅 효율과 정확성은 score에 의해 예상할 수 있는 것과 차이가 있을 수 있습니다. 또한 score가 낮은 gRNA들도 여전히 잘 작용할 가능성이 있습니다.
CRISPR 벡터를 VectorBuilder의 온라인 플랫폼에서 디자인할 때, 고객의 타겟 유전자에 대한 gRNA를 당사의 database에서 검색할 수 있는 옵션이 제공됩니다. 유전자의 이름을 넣으면 그 유전자에 대하여 당사의 database 내에 존재하는 guide RNA 디자인들의 상세한 정보를 보실 수 있습니다. 당사의 whole-genome gRNA database는 타겟 부위를 직접 찾고 분석할 필요 없이 원하시는 유전자를 위한 guide 서열을 쉽게 선택할 수 있게 함으로써, 널리 사용되는 다른 gRNA 디자인 툴이나 소프트웨어에 의해서 제공되는 장점들을 제공합니다.
VectorBuilder 온라인의 “정보 자료 - 학습 센터" 내에 CRISPR 실험을 위하여 성공적으로 실험 계획을 세우고, 실행하고, 문제를 해결하는 것을 도와 드릴 수 있는 위한 풍부한 자료들이 있습니다.
CRISPR 벡터 시스템에 대한 가이드를 읽어보시려면 여기를 눌러주시기 바랍니다.
CRISPR 구성 요소들에 대한 가이드를 읽어보시려면 여기를 눌러주시기 바랍니다.
자료
자주 묻는 질문
shRNA에 의한 knockdown과 nuclease (CRISPR 또는 TALEN) 에 의한 knockout 모두 세포 배양을 통해서 원하는 유전자의 loss-of-function 효과를 연구하기 위한 중요한 방법들입니다. 연구 목적을 위하여 어떤 방법이 가장 적절한지를 결정하기 위해서는 몇 가지 고려해야 할 사항들이 있습니다.
매커니즘
Knockdown 벡터
Knockdown 벡터는 세포 내에서 타겟 RNA의 절단을 유발하고 translation을 억제함으로써 타겟 RNA의 활성을 억제하는 short hairpin RNA (shRNA) 를 발현합니다. 따라서, shRNA knockdown 벡터는 원하는 유전자에 어떠한 DNA 수준의 서열 변화도 일으키지 않습니다.
shRNA knockdown 벡터에 대하여 더 보시려면 여기를 눌러주시기 바랍니다.
Knockout 벡터
CRISPR와 TALEN 둘 다 유전체 내의 특정한 타겟 부위를 절단하기 위하여 nuclease를 타겟 부위에 유도함으로써 작용합니다. 이러한 절단 부위는 세포내 기작에 의하여 비효율적으로 수리됨으로써, 그 부위에 짧은 서열이 삽입되거나 제거되면서 영구적인 변이가 생성되게 합니다. 이러한 변이 중 일부는 frame shift나 premature stop codon 등에 의하여 원하는 유전자의 loss-of-function이 일어나도록 합니다. 유전체 내에서 수 kb 이내의 가까운 거리에 위치하는 두 절단 부위가 동시에 타겟팅되는 경우, 그 사이의 서열이 제거되는 결과로 나타나기도 합니다.
CRISPR 벡터에 대하여 더 보시려면 여기를 눌러주시기 바랍니다.
효율
shRNA에 의한 knockdown으로는 절대로 타겟 유전자의 완전한 발현 억제가 되지 않습니다. 가장 효율적인 shRNA에 의해서도 어느 정도는 타겟 유전자의 발현이 유지됩니다. 반면에 CRISPR와 TALEN을 처리한 세포들의 경우, 일부의 세포들에서는 영구 변이를 통하여 원하는 유전자의 완전한 loss-of-function이 일어날 수 있습니다.
일관성 및 균일성
일반적으로 shRNA 벡터는 처리한 세포들 사이의 높은 균일성을 나타내며, 서로 다른 실험 결과에서도 일관된 결과를 보입니다. 반면에 CRISPR와 TALEN은 도입되는 변이의 무작위적인 특성에 의하여 처리한 세포들 사이의 높은 비균일성을 나타냅니다. 하나의 세포 내에서 원하는 유전자의 완전한 knockout을 위해서는 세포 내에 존재하는 해당 유전자의 모든 copy를 knockout해야 합니다. 정상 세포들은 X- 또는 Y-염색체에 있는 유전자들을 제외하고는 모두 2 copy씩 가지고 있으며, 암세포들은 2 copy 이상을 가질 수 있어서, 전체 세포들 중 매우 작은 부분만이 완전한 knockout이 일어난 세포들일 수 있습니다. 이러한 이유로, nuclease에 의한 knockout 실험에서는 원하는 유전자의 모든 copy들이 knockout된 subset을 확인하기 위하여 sequencing에 의하여 clone들을 screening해야 합니다.
Off-target 효과
Off-target 효과는 shRNA에 의한 knockdown과 nuclease에 의한 knockout 둘 다에서 보고되었습니다. Off-target의 표현형은 같은 유전자를 타겟팅하는 다른 여러 개의 shRNA를 사용함으로써 추정할 수 있습니다. 여러 개의 다른 shRNA에 의하여 일관되게 나타나는 knockdown 표현형이 있으면, 그 결과가 off-target 효과에 의한 것이 아니라고 할 수 있습니다. CRISPR와 TALEN에 의한 knockout에서는 off-target 변이에 의한 표현형을 설명하기 위해서는 여러 개의 loss-of-function을 초래하는 변이를 포함하는 clone들을 분석해야 합니다. 또한, bioinformatics에 의하여 발견한 잠재적인 off-target 부위들에 대한 변이 여부를 확인하기 위하여, sequencing으로 clone들을 확인하여야 합니다.
CRISPR와 TALEN 시스템은 둘 다 배양된 세포와 동물 모델에서 유전체 편집을 위하여 사용되어 왔습니다. 두 시스템 모두 유전자 knockout 및 point mutation 이나 insertion을 위한 knockin에 사용될 수 있지만, 이 두 시스템은 여러 가지 면에서 다르고 각각의 장단점을 지니고 있습니다.
매커니즘
CRISPR
CRISPR 시스템은 site-specific guide RNA (gRNA)를 사용하여 Cas9 nuclease를 유전체 내의 타겟 부위로 유도하여 DNA 절단이 일어나도록 합니다. 타겟 서열은 일반적으로 20 bp 정도의 길이를 가지며, 소수의 염기가 불일치하는 다른 부위의 서열도 인식되고 절단될 수 있습니다.
CRISPR 벡터에 대하여 더 보시려면 여기를 눌러주시기 바랍니다.
TALEN
TALEN 시스템은 한 쌍의 키메라 단백질을 이용하는데, TAL effector의 DNA-binding domain (특정한 서열을 인식함) 을 FokI 제한효소의 nuclease domain과 결합하여 구성됩니다. 유전체 내에서 14-20 bp의 spacer 서열 (절단 부위) 양쪽으로 18 bp 정도의 길이를 갖는 한 쌍의 타겟 부위에 결합하도록, 한 쌍의 TALEN 단백질을 디자인합니다. DNA에 결합하면, 한 쌍의 TALEN 단백질의 Fokl nuclease domain들이 dimer를 형성하여, 두 타겟 부위 사이에 존재하는 spacer 서열의 DNA를 절단하게 됩니다.
효율
CRISPR와 TALEN에 의한 유전체 편집은 둘 다 좋은 효율을 보여주지만, 응용 분야, 생물종 및 세포 유형에 따라 효율에 차이가 있습니다. 일반적으로 CRISPR가 TALEN에 비하여 세포내로 전달되고 DNA 절단을 유도하는데 더 효율적입니다.
Off-target 효과
CRISPR gRNA는 약 20 bp 길이의 서열을 타겟팅하는 반면에, TALEN 한 쌍은 36 bp의 타겟 서열에 결합을 합니다. 또한 Cas9/gRNA complex는 TALEN 보다 큰 5 bp 까지의 mismatch를 허용합니다. 따라서, TALEN에 의한 절단은 CRISPR에 의한 것보다 더 높은 서열 특이성를 가지고 있으며, 유전체 내에서 TALEN에 의한 off-target 부위의 절단은 거의 일어날 수 없습니다. 반면에, 세포주들에서 CRISPR에 의한 off-target 효과가 보고되어 왔으며, CRISPR knockout 마우스에서 분석한 결과로는 in vivo에서는 더 낮은 빈도로 off-target 효과가 나타나는 것으로 보입니다. 최근에 개발되고 있는 CRISPR 시스템들은 상당히 향상된 CRISPR에 의한 서열 특이성을 지니고 있습니다. Cas9 nickase (Cas9_D10A, Cas9_H840ACas9 과 같이 하나의 catalytic nuclease domain만을 갖는 Cas9 변이) 와 이중 gRNA를 사용하는 경우, 타겟 부위 가까이에 생기는 두 개의 single-strand DNA nick들에 의하여 double-nick DSB (double-strand break)가 생성되면서 수리됩니다. 이러한 디자인에 의하면, 이중 gRNA에 의하여 타겟 부위가 40 bp 정도까지 확장될 수 있어서 off-target 효과를 최소화할 수 있습니다.
타겟 부위의 조건
TALEN은 유전체 내의 어떠한 서열도 타겟으로 할 수 있습니다. 반면에, CRISPR 타겟 부위는 gRNA 타겟 서열의 3' end에 존재해야 하는 PAM 서열 (일반적으로 NGG) 을 필요로 하기 때문에 타겟 서열 선택에 제한이 있습니다. 유전자 knockout의 경우에는 유전자 내의 어느 부분이든 절단되면 잠재적으로는 효과적이기 때문에 문제가 되지 않지만, 유전자 내의 정확한 부위에 변이를 일으키거나 서열을 삽입해야 하는 경우 어려움이 있을 수 있습니다. CRISPR를 사용하여 유전체 내의 특정한 부위에 정밀한 편집을 하기 위해서는 gRNA, Cas9과 함께, 타겟 서열의 바로 앞과 뒤에 homology arm을 추가한 homologous recombination donor 벡터나 길이가 긴 oligo를 전달함으로써 타겟 부위에서 HDR (homology directed repair)에 의한 DNA repair가 일어나도록 할 수 있습니다.
고객의 homologous recombination donor 벡터를 온라인에서 디자인하시려면 여기를 눌러주시기 바랍니다.
기술적인 어려움
기술적인 단순성의 측면에서는 여러 가지로 CRISPR가 TALEN보다 낫습니다. 첫째, 벡터 제작에 있어서 CRISPR 시스템은 Cas9/gRNA complex에 의한 타겟팅이 단순히 RNA/DNA hybridization에 의하여 일어나기 때문에 짧은 gRNA만 클로닝을 하면 됩니다. 반면에 TALEN 시스템은 각각의 단백질-DNA 상호작용을 위한 고유한 TAL DNA-binding domain을 다시 제작해야 합니다. 따라서, 각각의 타겟 부위마다 두 개의 벡터를 필요로 하는 TALEN에 비하여, gRNA는 디자인과 클로닝 측면에서 더 적은 비용으로 더 쉽게 제작할 수 있습니다. 둘째, 마우스 배아에 injection 하는 것과 같은 몇가지 응용 분야에서 Cas9 단백질과 gRNA는 direct injection에 의하여 효율적으로 전달이 될 수 있지만, TALEN은 그렇지 않습니다. 세째, 수천의 서로 다른 gRNA를 발현하는 CRISPR screening library는 high-throughput 방식으로 쉽게 제작될 수 있기 때문에, CRISPR는 genetic screening 실험을 위한 엄청난 다양성을 제공합니다.
CRISPR에 의한 유전체 편집을 위하여 Cas9 nuclease는 특정한 부위에 대한 guide RNA (gRNA)에 의하여 유도되어 DNA를 절단부위를 생성합니다. 대부분의 경우에 단순한 유전자 knockout을 위하여 하나의 gRNA를 Cas9과 사용하여 double-strand break (DSB)를 생성하고, DSB가 non-homologous end joining (NHEJ)에 의하여 비효율적으로 수리된 결과로 작은 insertion 또는 deletion이 일어나면서 영구적인 변이가 도입됩니다. 이러한 영구 변이들 중 일부에서는 frame-shift나 premature stop codon 등에 의하여 원하는 유전자의 loss-of-function이 일어나게 됩니다.
하나의 타겟 부위에 대하여 두 상반된 strand를 절단하기 위하여 Cas9_D10A nickase를 사용하는 경우에 이중 gRNA가 사용될 수 있습니다. 이 방법에서는 nickase enzyme이 각각의 strand에 각각의 gRNA에 의하여 유도되면서 single strand cut을 만들어, 타겟 부위에 DSB를 생성하게 됩니다. DSB를 생성하기 위해서는 두 gRNA 모두에 의해서 타겟팅되어야 하기 깨문에, 일반적으로 이 방법에 의하면 CRISPR/Cas9 발현에 의한 off-target 효과가 감소하게 됩니다.
이중 gRNAs 는 Cas9_D10A nickase와 외래의 donor DNA template를 사용하여 원하는 유전자에 특정한 서열 변화를 도입하는 knockin을 위하여 사용될 수 있습니다. 이 방법에서는 원하는 변이 부위 주위를 타겟으로 하는 두 개의 gRNA에 의하여 상반된 두 strand를 절단하고, homology-directed repair (HDR) pathway에 의하여 외래의 donor template를 이용하여 절단 부위를 수리하게 됩니다.